PyQInt: a Python package for evaluating Gaussian integrals and performing electronic structure calculations

PyQInt is a Python package for calculating one- and two-electron integrals as encountered in electronic structure calculations. Since integral evaluation can be quite computationally intensive, the evaluation is programmed in C++ and connected to Python using Cython.
PyQInt mainly serves as an educational package to teach students how to perform (simple) electronic structure calculations wherein the most difficult task, i.e. the integral evaluation, is already encapsulated in a handy set of routines. With PyQInt, the student can for example build their own Hartree-Fock routine. Some common electronic structure routine, most notably the Hartree-Fock algorithm, is also readily available.
PyQInt offers supporting scripts for facile visualization of result such as producing contour plots for the molecular orbitals. Below, an example is shown for the molecular orbitals of the CO molecule.
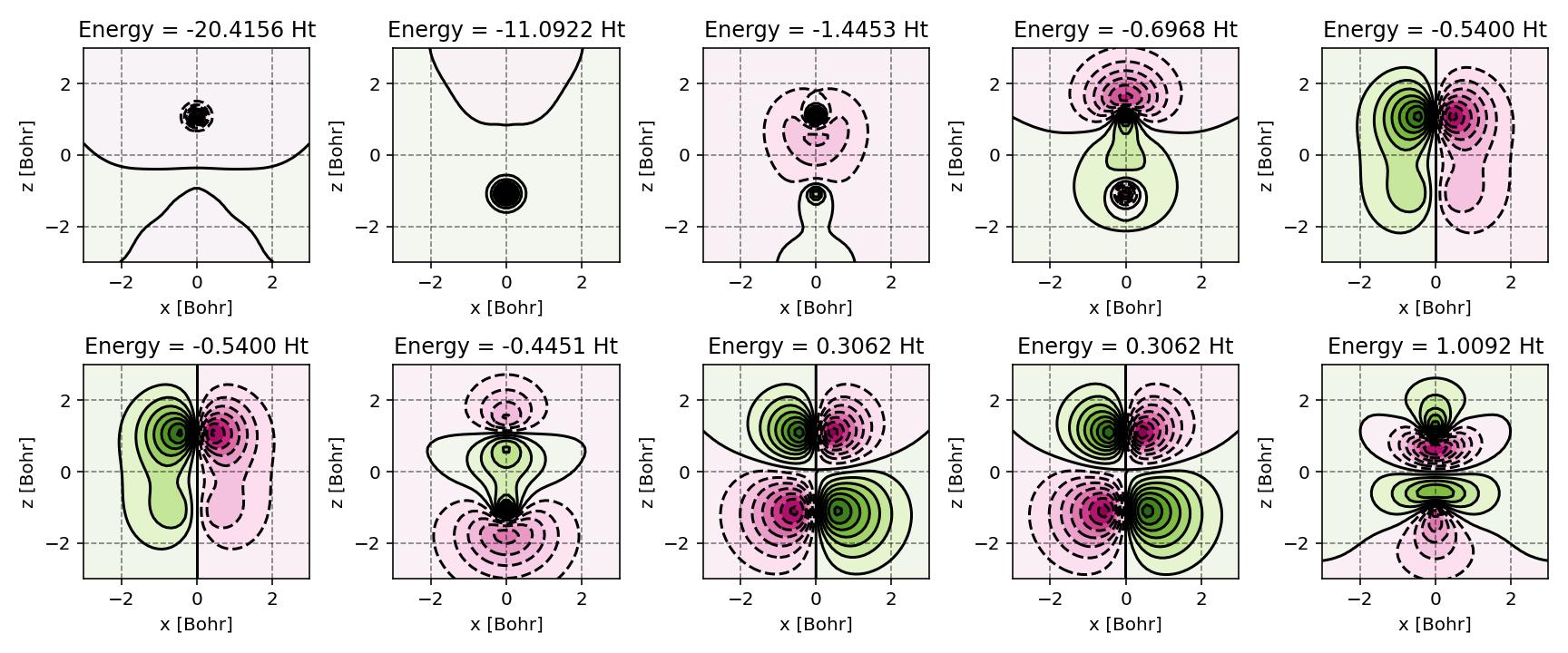
Canonical molecular orbitals of CO visualized using contour plots.
PyQInt not only supports calculation of the canonical molecular orbitals via the (restricted) Hartee-Fock procedure, but can also be used to construct the localized molecular orbitals which is relevant for showing the similarity between modern electronic structure methods and classical Lewis theory. In the image below, one can observe the canonical molecular orbitals for the CO molecule as well as the localized molecular orbitals.
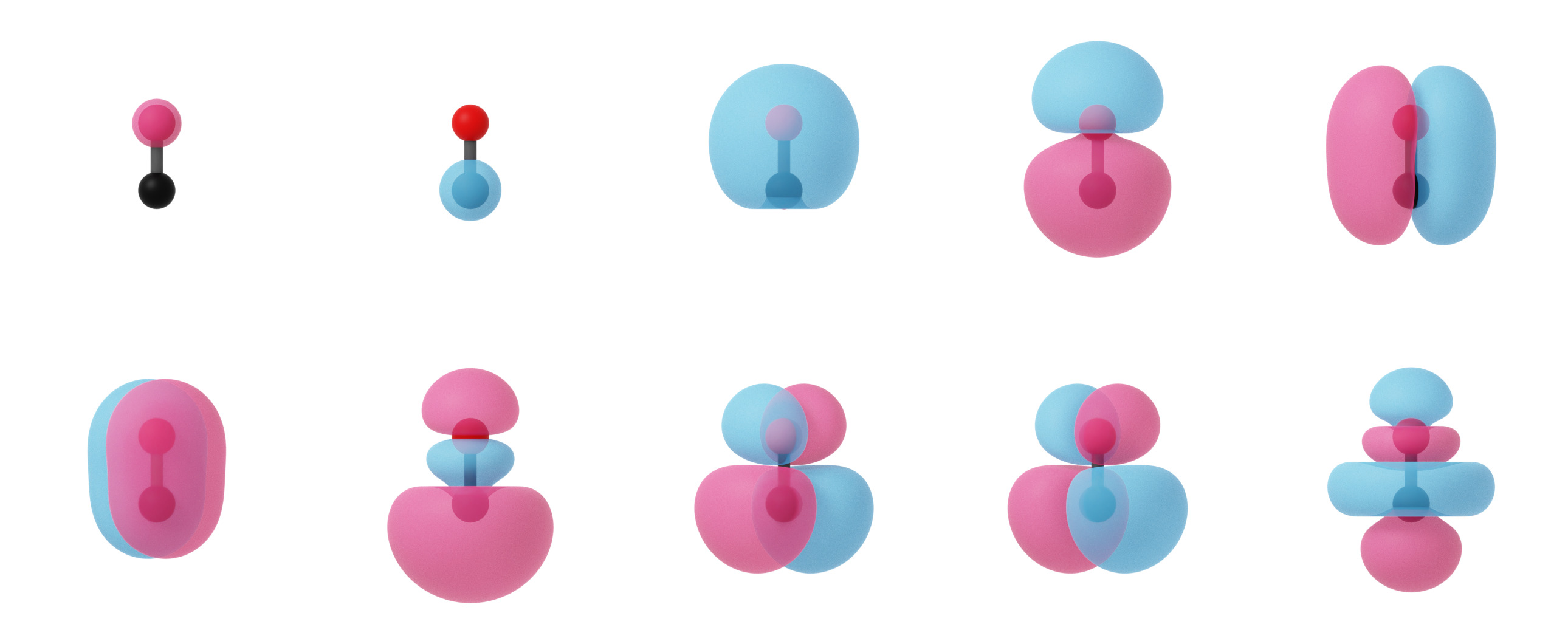
Canonical molecular orbitals of CO visualized using isosurfaces with an isovalue of +/-0.03.
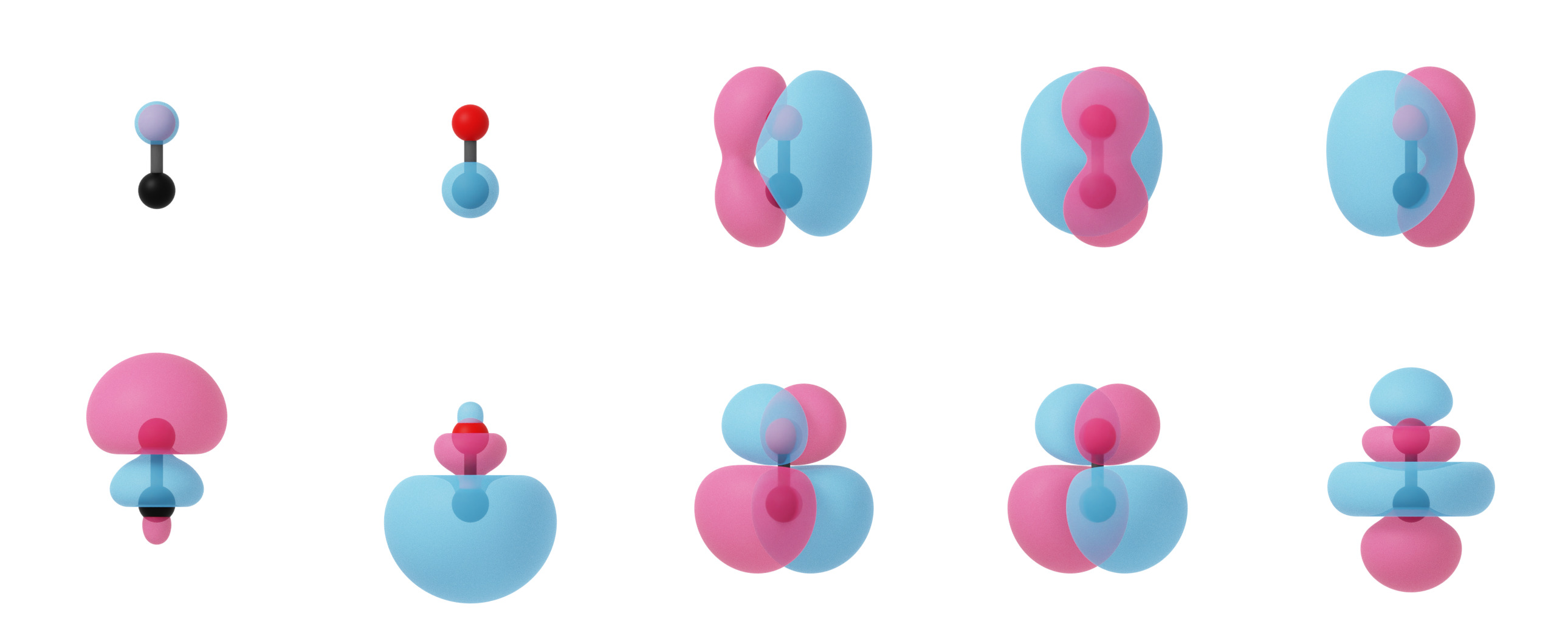
Localized molecular orbitals of CO visualized using isosurfaces with an isovalue of +/-0.03. Note that the localization procedure has only been applied to the occupied molecular orbitals. Observe that the localized orbitals contain a triple-degenerate state corresponding to the triple bond and two lone pairs for C and O.
PyQInt has been developed at the Eindhoven University of Technology, Netherlands. PyQInt and its development are hosted on github. Bugs and feature requests are ideally submitted via the github issue tracker.
Contents: